Clean Code
“If you can't even clean up your own room, who are you to give advice to the world?” Jordan B. Peterson
Our main goal is to use causal molecular knowledge and signaling activity data to build and parameterize Boolean models to represent specific cancer cell systems, and study their behavior in the presence of in-silico drug perturbations. As such, the importance of the underlying scientific software that enables these computational tasks is unquestionable. Such software needs to satisfy a list of requirements pertaining to its suitability for practical use [92]. Such practices ensure that the software does what it is intended to do and its produced scientific simulations can be used to inform decision-making for clinical applications. In other words, for the scientific results of the simulations to be actionable and trustworthy [93], the following software requirements are not just optional, but rather a necessity. At first, challenges related to automation, efficiency and optimization in terms of the computational resources necessary to perform the simulations and analyses, need to be properly addressed. To promote collaboration and allow others to study, use and further develop the software, the respective codebase needs to be open sourced [94,95]. Standard formats for input and output should be supported, as well as standard libraries for common programming tasks, enabling the effortless integration with related software. Also, sufficient documentation needs to be provided, containing installation guidelines and explanations for the various configurations used in the simulations [96]. Simple examples of use and related tutorials should be part of such documentation as well [97]. The usability of the software can also be increased by applying better programming architecture and design principles (e.g. writing modular code), which also makes the software easier to test, extend and verify. Making the results of the simulations verifiable (i.e. by ensuring that the algorithmic procedures and the model equations are correctly coded),6 will increase their reproducibility, further supporting the aforementioned goals [98]. All in all, there can only be gains if the code is clean and properly taken care of [99].
“The standard library saves programmers from having to reinvent the wheel.” Bjarne Stroustrup

In Figure 5 we present an overview diagram of the DrugLogics software pipeline. This pipeline represents a computational software system aiming to assist in the identification of synergistic drug combinations. The pipeline’s two main modules, Gitsbe (Generic Interactions To Specific Boolean Equations) and Drabme (Drug Response Analysis to Boolean Model Ensembles), are both written in Java. Gitsbe, using as input a PKN and a signaling activity profile for some of the key entities in the input network, creates Boolean models based on a genetic algorithm approach and calibrates them to best fit the training signaling activity data. Boolean models with higher fitness have fixpoint attractors whose node states better match the binary signaling activities. Calibration refers to changes in the Boolean equations to produce such high fitness models. These changes can simply be variations in the parameterization such as mutations in the Boolean rules (e.g. a logical OR becomes a logical AND and vice-versa) or topological changes, such as the addition or removal of a regulator’s effect on a particular target. Thus the Boolean models in Gitsbe’s produced ensemble are parameterized differently, but are all essentially approximate representations of the same biological system. These variant Boolean models are then used as input to Drabme, to test the effect of several perturbations from a given drug panel and quantify their combinatorial interaction patterns using well-known synergy frameworks (namely HSA [100] and Bliss [101]). Leveraging the wisdom of the crowds [102,103] using Gitsbe’s models, Drabme’s output predictions contribute in reducing the exponentially large drug space that is associated with combinatorial treatments for the targeted therapy of cancer, providing a list of candidate combinations to test in the lab, before a viable solution is found for a patient.
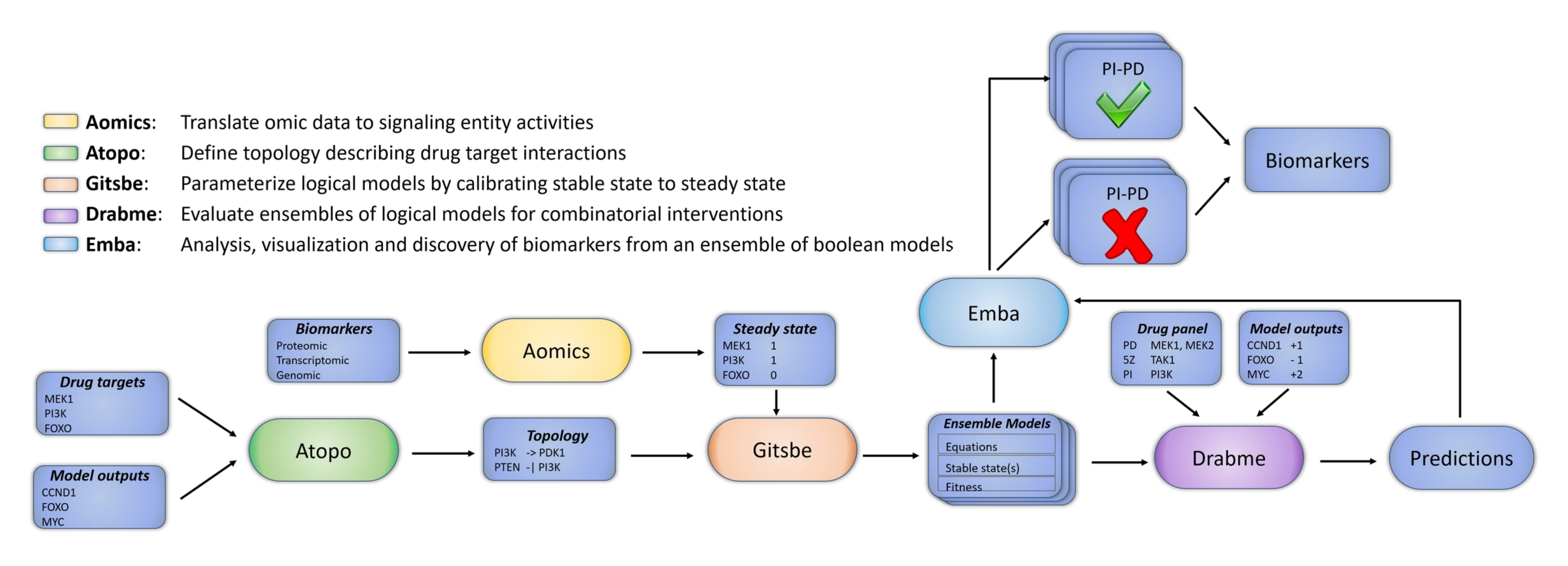
Figure 5: The DrugLogics software pipeline. A series of connected modules that build a regulatory topology incorporating specific drug targets, parameterize Boolean models to a specific cancer signaling profile assembled from various omics data and simulate drug combination perturbations. The output models and their predictions can be further analyzed to explain the difference between phenotypes and thus identify biomarkers that make a particular drug combination synergistic.
The development of Drabme and Gitsbe was part of the latest stages of a previous research thesis and it was mostly exploratory work [104].
To lay the groundwork for the analyses and investigations of the papers included in this thesis, additional software development needed to be done.
We started by refactoring the source code to increase its readability, maintainability and extensibility.
Java classes were restructured, variables and functions were renamed to reflect current best programming practices, code written from scratch for common tasks such as string manipulation was replaced with reusable Java components (e.g. using libraries from Apache Commons [105]) and the Maven project management tool [106] was used to enable easier source compilation, installation and packaging (all the Java code was bundled in a single compressed file for ease of use).
To increase interoperability of our software, we used the Java library BioLQM [107] to enable the export of the produced Gitsbe models to standardized formats in the logical modeling community, such as GINML (used in GINsim, a software tool enabling the definition, analysis and simulation of regulatory graphs based on the logical formalism [108]), SBML-qual (a standard designed for the representation of multivalued qualitative models of biological networks [109]) and BoolNet (file format of the models built with the BoolNet R package, used for the simulation and analysis of Boolean networks [110]).
For the calculation of fixpoint attractors, an external scripting-based tool was used by default [111].
We added a built-in, integrated Java solution using BioLQM, which apart from fixpoints can also identify minimal trapspaces, a generic type of attractor that allows for a deeper exploration of dynamics.
... and this is how happy I was when I finally cleaned up the DrugLogics code:

A practical outcome of these efforts was that the code became more modular, enabling the addition of software tests using the JUnit5 framework [112] and specialized libraries [113,114].
With more tests, hidden or otherwise impossible to pinpoint bugs were identified and fixed.
Some of these were critical for the validity of the output findings, since they related to how changes in the parameterization or topology were encoded in the software equivalent of a Boolean model’s equations.
Moreover, it became much easier to add new features to the software, e.g. we supported the execution of parallel simulations in Gitsbe (a significant performance optimization) and incorporated a new synergy framework for the identification of synergistic drug combinations in Drabme (Bliss).
Gitsbe’s simulation is the core computational process that produces the “best-fit” models resulting from the evolutionary approach of the genetic algorithms.
Selecting a few of those best-fit models at the end of each simulation and executing multiple simulations in parallel, each one associated with a different random seed number, is what generates a reproducible list of Boolean models for use in Drabme (or other software if models are exported to standard formats).
Lastly, we shared publicly the developed modules in GitHub7 and built an extensive online documentation using the R package bookdown [115] to gather all related information in one place with regard to the software’s configuration parameters, the mathematical calculations used, installation instructions and examples of use.8
This online documentation became a central virtual hub, providing information on all the software modules in the pipeline.
One of these modules was druglogics-synergy, a Java package used to serially execute Gitsbe and Drabme in one go, and which was employed for the simulations of Paper 3.
Even a small bug can have severe cascading effects...

With all the main code in proper order, we could start investigating the outputs of our software and assess the quality of the produced drug synergy predictions.
The results of these efforts, along with biologically-relevant mechanistic insights derived from further analyzing the simulation data and performing various investigations, are analytically presented in Paper 3, and in even more detail at the ags-paper repository,9 which also includes reproducibility guidelines.
In the following paragraphs we are going to briefly explain the input data and software that either needed to be in place before we started the experimentation with the in-silico simulations or was built to help further analyze the output Boolean models from Gitsbe and Drabme’s synergy predictions.
To begin with, we had to choose a reference drug combination dataset to compare our predictions against. The argument here is to use a dataset that you know very well so that you can first experiment with the software and its configurations, and only later test your predictions on other published (and potentially larger) datasets. Therefore, we chose the Flobak et al. (2019) dataset, with a total of 153 combinations of 18 targeted drugs, involving measurements across 8 cancer cell lines [116]. Next, we needed to assess in a computational manner which of the drug combinations in the reference dataset (per cell line) are synergistic and which are not. Our first attempt was to manually check the output growth curves and derive a majority-assessed gold standard (so it was more of a curative group effort). We continued by performing a thorough analysis using the CImbinator tool [117] and established a methodology which computed the synergy classification of the reference dataset that best matched our internal curation efforts to call synergy.10 All in all, we had a reference dataset and a list of drug combinations designated as synergistic from it, ready to be used to evaluate Drabme’s predictions on the same dataset.
On another front, we also needed a PKN suitable for our analysis. In particular, the targets of the drugs used in the reference dataset should be entities in the network, so as to enable the simulation of drug perturbations in Gitsbe’s derived Boolean models. Moreover, several of the most important pathways in cancer cell biology (e.g. PI3K, ERK and TGFB signaling [118]) would have to be included as well. A PKN that fits all the above characteristics was curated within our research group and refined throughout many years, resulting in the topology that was used for the simulations of Paper 3 (CASCADE).11 In addition, proper signaling data was needed to train the Gitsbe models to a cancer proliferating phenotype. For that purpose, we used the literature curated activity profile for a set of nodes in CASCADE, which was the result of a previous research effort [90]. This activity profile concerns only one of the cell lines in the reference dataset, namely the gastric adenocarcinoma cell line (AGS). In summation, by employing a curated topology and training data from only one cell line, we could focus more on the model parameterization aspects of our software, the performance assessment of the synergy predictions and the rest of the investigations of Paper 3.
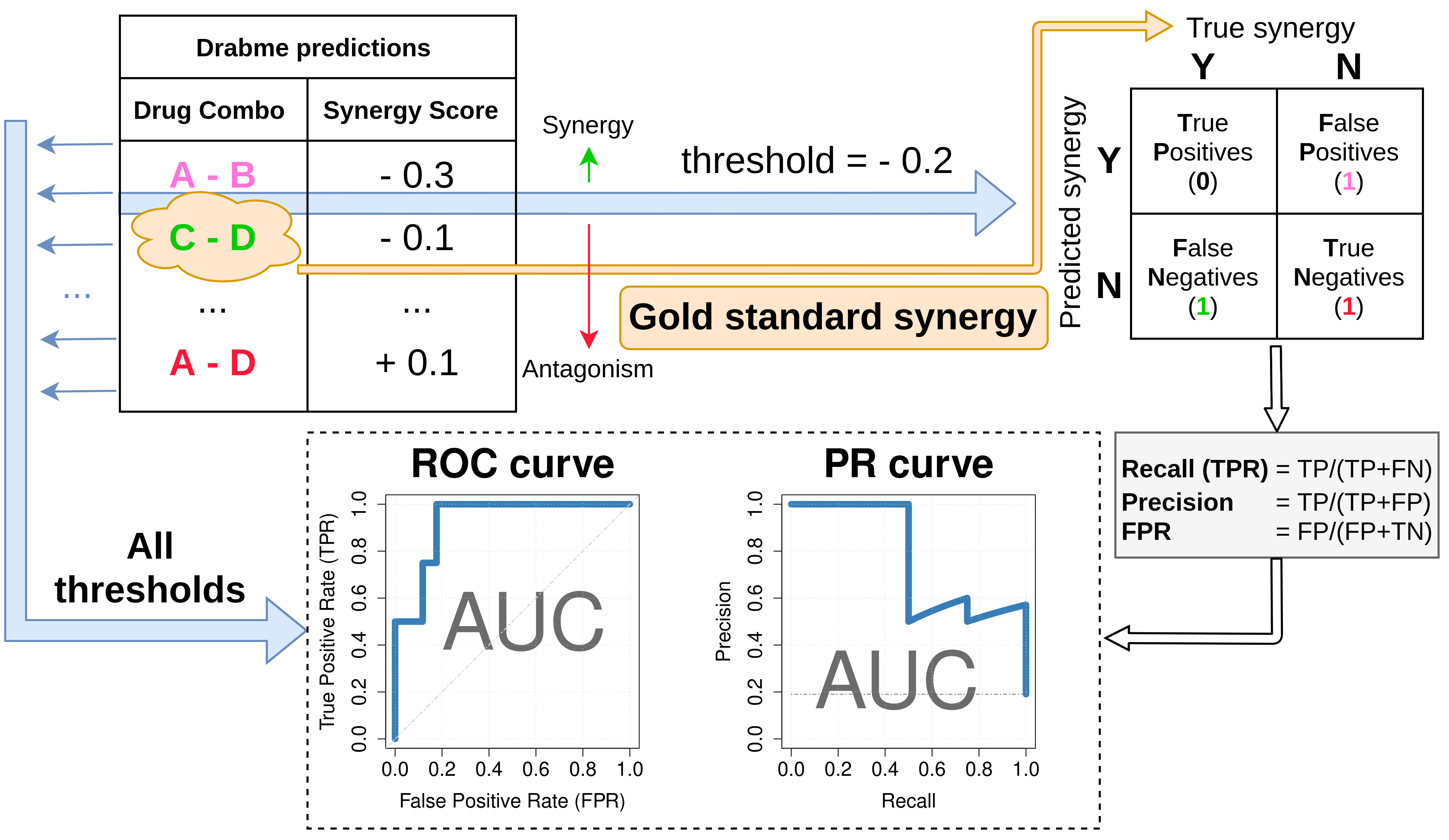
Figure 6: Performance assessment of Drabme’s drug synergy predictions. Predicted synergy scores are sorted from synergistic to antagonistic and compared to a gold standard synergy set for several possible thresholds. Each synergy threshold can be used to construct a confusion matrix, from which standard metrics are calculated, such as the number of True Positive (TP) and False Positive (FP) predictions, precision and recall, etc. Visualizing several of these metrics across all thresholds in the Receiver Operating Characteristic (ROC) and Precision Recall (PR) curves, enables the calculation of the Area Under the Curve (AUC), which is a performance score indicating how good the synergy classification method is.
While exploring various configuration options for the Gitsbe and Drabme simulations,12 we needed a tool to quickly assess their effect on the pipeline’s performance and see which were the most important to tune.
The pipeline’s performance here refers to Drabme’s output synergy predictions (continuous scores, each one for a different drug combination, ranging from negative and more synergistic values, to positive and more antagonistic values), validated against the computationally derived set of synergistic drug combinations for the AGS cell line (gold standard).
This is a typical binary classification problem, where an imaginary threshold scans the range of Drabme’s predicted synergy scores to derive various performance scores.
Specifically, each threshold demarcates the synergistic drug combinations from the antagonistic ones, based on the output prediction scores.
By comparing these assessments with what we consider as the actual truth, i.e. the gold standard synergy set, a confusion matrix can be constructed and several threshold-specific measures calculated (Figure 6).
By accounting for every such threshold, ranging from a very strict (every prediction is declared as antagonistic) to a very relaxed one (every prediction is declared as synergistic), we can visualize Drabme’s classifier performance with the Receiver Operating Characteristic (ROC) [119] and Precision Recall (PR) [120] curves, which are both well-known diagnostic tools for binary classification problems.
In particular, they allow for a more broad, threshold-agnostic performance measurement, which is the Area Under the Curve (AUC).
An AUC score of 1 is considered as the perfect classification, which in our case would happen if the top most negative synergy scores were exactly the ones corresponding to the drug combinations that represent the gold standard set for the AGS cell line.
The aforementioned diagnostic curves also enable the calculation of “optimal” cutpoints, e.g. thresholds that maximize or minimize certain criteria, an example of the latter being the distance from the point of perfect classification [121].
Based on this theoretical framework, we developed the R shiny app [122] druglogics-roc to automatically parse Drabme’s output files and produce a table [123] with the confusion matrix values per synergy threshold, along with interactive plots of the ROC (using the R package plotly [124]) and PR curves (using the R package PRROC [125]) and their respective AUC scores.
This app is now part of the DrugLogics software suite,13 facilitating the visualization of the pipeline’s performance.
The Boolean model ensembles produced by Gitsbe were a unique source for further data analyses.
Since such an ensemble contains a large variety of models, all representing the same biological system, we investigated how these models differ in terms of each network node’s activity (in the respective attractor) and parameterization.
Moreover, we were interested in how these types of model differences translate to variations in prediction performance, i.e. make some models predict specific drug combinations as synergistic or not.
By assigning Gitsbe models into different classes based on their individual prediction performance, we could identify nodes that were relatively more active (or inhibited) in the upper tier models compared to the lower tier models of the performance hierarchy.
For example, we verified across many analyses14 that the ERK node of the CASCADE signaling network was particularly overexpressed in the models that predicted most of the gold standard synergies in the AGS cell line from the reference drug combination dataset.
This was an interesting finding, since knowledge gathered from the scientific literature indicates conflicting measurements of ERK activity in AGS cells [90] and so our modeling results, upon further analysis, have the potential of providing useful information related to the studied biological system (this particular result is also shown using a different methodology in Paper 3).
In addition to the activity-based analyses, we also explored differences with respect to the Boolean model parameterization, i.e. if higher performance models tend to have specific logical operators (or not) in some equations.
This also motivated us to study how the diversity in particular Boolean rule assignments in the different model classes translates to the respective target nodes’ activity (the link between node parameterization and activity was further investigated in Paper 5).
To enable all the aforementioned investigations, we began writing functions in several scripts while getting familiar with the world of professional software development in R [126].
In the end, we spent considerable effort to organize all these functions into a single, clean, modularized and tested codebase, with the purpose to fill in a niche for data analysis-oriented software that performs auxiliary automated analyses on Boolean model datasets.
The result was the creation of the emba R package and its addition to the DrugLogics software suite (Paper 4).